A disease that affects approximately one out of a million people, Familial Lipoprotein Lipase Deficiency (also known as chylomicronemia, or LPLD for short), is an autosomal recessive disease caused by the mutation of the LPL gene that is responsible for coding lipoprotein lipase. Interestingly, it is more common in Quebec, Canada due to consanguinity (blood relations).
Lipoprotein lipase is an enzyme that is activated by apolipoprotein C-II, a cofactor, to hydrolyse fats from two types of lipoproteins - chylomicrons and low density lipoproteins (VLDLs). Chylomicrons are responsible for transporting fat from the intestine to the bloodstream while VLDLs circulate in the blood, carrying fat and cholesterol from the liver to other tissues in the body.
Under normal circumstances, the fats from such lipoproteins are broken down to release energy or to be stored in adipose tissue by activated lipoprotein lipase.
However with LPLD, the function of lipoprotein lipase is impaired, chylomicrons and VLDLs accumulate, which is followed by the accumulation of triglycerides.
What causes this disease?

LPL gene is located at the short arm of chromosome 8, position 22 (Cytogenic location of 8p22)
While it is discovered that over 220 mutations in the LPL cause familial lipoprotein lipase deficiency, point mutation of amino acid glycine to glutamic acid at position 188 in the enzyme is the most common. While glycine and glutamic acid are polar molecules, glycine is uncharged while glutamic acid is negatively charged, thus causing a change in the shape of the enzyme. As such, the function of lipoprotein lipase is reduced or lost, causing an accumulation of fat-containing chylomicrons in the blood stream. Many symptoms and indications of familial lipoprotein lipase deficiency will follow.
Biochemistry Process
Before we touch on the symptoms of LPLD, let us go further into the functions of liproteins (chylomicrons and low-density lipoprotein) and their involvement in lipid metabolism.
What is a lipoprotein?
A lipoprotein is a molecule that consists of a lipid bound to a protein (apoprotein), where the protein serves to emulsify the lipid so that it can move in and out of the cell via water.
The detailed functions of liprotein are split into two pathways - exogenous and endogenous - and depends on whether the lipid bound to the apoprotein is composed of dietary lipids or is synthesized in the liver.
Chylomicrons are lipoprotein that are function in the exogenous pathway while very low-density liproteins (VLDLs) function in the endogenous pathway.
The composition of chylomicrons and VLDLs play an important role in their function, and are as follows:
Chylomicrons
|
VLDLs
|
|
Protein
|
1%
|
10%
|
Triglyceride
|
88%
|
56%
|
Cholesterol
|
1%
|
8%
|
Cholesterol esters
|
3%
|
15%
|
The difference between cholesterol and cholesterol ester is shown below:

While cholesterol contains an alcohol group, cholesterol ester contains an ester group, thus is more hydrophobic than normal cholesterol.
Chylomicrons

Chylomicrons contain two types of apoprotein - integral and peripheral. Intergral proteins are apoA and apoB while peripheral proteins are apoC and apoE.
Steps of exogenous pathway:
1. Fats are emulsified by bile into smaller fat droplets in the small intestine.
2. Pancreatic lipase, colipase and bicarbonate are released into the small intestine. Bicarbonate neutralises stomach acid, colipase binds to the emulsified fats and pancreatic lipase breaks them down into free fatty acid and 2-monoacylglycerol.
3. The fatty acids and 2-monoacylglycerol are absorbed into enterocytes (intestinal epithelial cells)
4. After absorption, they recombine to form triglycerides.
5. Recombined triglycerides and cholesterol packaged into nascent chylomicrons which are synthesized by apoB48, an integral protein.
6. Chylomicrons released from basolateral site of enterocytes into the lacteal which is part of lymphatic system - a process that relies on apoB48.
7. Travel through lymphatic system and enter thoracic duct, where it gets drained into the blood stream.
8. In the blood stream, HDL (high density lipoprotein) donates apolipoprotein C-II (apoCII) and apolipoprotein E (apoE) to nascent chylomicron. The chylomicron is then considered matured.
9. Chylomicrons travel down blood vessels and can either go into liver or adipose tissue.
10a) If chylomicrons travel to the liver, apoE of chylomicrons will bind to the LDL receptor of the liver cells. Chylomicrons will be taken up by the liver and triglycerides will be used.
10b) If chylomicrons travel to the adipose tissue, apoC-II activates the lipoprotein lipase present on the endothelial cells lining the blood vessels. Lipoprotein lipase will then cleave triglycerides into fatty acids and glycerol which will absorbed by adipose and muscle tissue for energy and storage. When the triglyceride level in chylomicrons is reduced to 20%, chylomicron loses apoC and becomes a chylomicron remnant. It goes to the liver and apoE will bind to the chylomicron remnant receptor in the liver cell. Endocytosis occurs and chylomicron remnants are then hydrolzed within lysosomes, releasing the remaining glycerol and fatty acids to be used for energy or stored for later use.
Very Low-Density Lipoprotein (VLDL)
Steps of Endogenous pathways:
1. Triglyceride and cholesterol, together with apolipoprotein B-100 are assembled in the liver to form nascent VLDL.
2. Nascent VLDL depends on apolipoprotein B-100 to be released into the bloodstream.
3. HDL donates ApoC-II and ApoE to nascent VLDL, making them mature.
4. VLDL encounters LPL on the endothelial cells. ApoC-II activatse LPL, causing hydrolysis of VLDL and releases glycerol and fatty acids that are absorbed by the peripheral tissues (such as adipose and muscle tissue) from blood.
5. The mature VLDL now becomes VLDL remnants or Intermediate Density Lipoproteins (IDLs).
6. The apoE of IDLs binds to the remnant receptor of the liver and is absorbed. ApoE of IDLs can also bind to the LDL receptor of the liver, causing absorption via endocytosis to the liver.
7. VLDL remnants can also be hydrolyzed by hepatic triglyceride lipase (HTGL) in the liver, releasing glycerol and fatty acids. VLDL remnants now become Low Density Lipoproteins (LDL) that contain a relatively high cholesterol content and are smaller and denser than IDLs.
8. LDL is circulated and apoB-100 binds to the LDL receptor which can be found in hepatic and non-hepatic cells, causing absorption of LDL via endocytosis.
In the liver, LDL is converted into bile acids which are secreted into the intestine.
In non-hepatic cells, LDL is hydrolzyed by lysosomes, releasing lipids which are mostly cholesterol into the cells. The cholesterol will be used for cell membrane and steroid hormone synthesis.
Plasma LDL concentration is regulated by LDL receptor activity, which in turn, is regulated by several mechanisms. A few examples of such mechanisms are decreasing synthesis of HMG-CoA which controls cholesterol synthesis (hydroxy-3-methyglutaryl coenzyme A reductase), suprressing synthesis of new LDL receptors in the cells, activating the enzyme acyl-coenzyme A cholesterol acltransferase which helps esterify free cholesterol to cholesterol ester.
Steps of Endogenous pathways:
1. Triglyceride and cholesterol, together with apolipoprotein B-100 are assembled in the liver to form nascent VLDL.
2. Nascent VLDL depends on apolipoprotein B-100 to be released into the bloodstream.
3. HDL donates ApoC-II and ApoE to nascent VLDL, making them mature.
4. VLDL encounters LPL on the endothelial cells. ApoC-II activatse LPL, causing hydrolysis of VLDL and releases glycerol and fatty acids that are absorbed by the peripheral tissues (such as adipose and muscle tissue) from blood.
5. The mature VLDL now becomes VLDL remnants or Intermediate Density Lipoproteins (IDLs).
6. The apoE of IDLs binds to the remnant receptor of the liver and is absorbed. ApoE of IDLs can also bind to the LDL receptor of the liver, causing absorption via endocytosis to the liver.
7. VLDL remnants can also be hydrolyzed by hepatic triglyceride lipase (HTGL) in the liver, releasing glycerol and fatty acids. VLDL remnants now become Low Density Lipoproteins (LDL) that contain a relatively high cholesterol content and are smaller and denser than IDLs.
8. LDL is circulated and apoB-100 binds to the LDL receptor which can be found in hepatic and non-hepatic cells, causing absorption of LDL via endocytosis.
In the liver, LDL is converted into bile acids which are secreted into the intestine.
In non-hepatic cells, LDL is hydrolzyed by lysosomes, releasing lipids which are mostly cholesterol into the cells. The cholesterol will be used for cell membrane and steroid hormone synthesis.
Plasma LDL concentration is regulated by LDL receptor activity, which in turn, is regulated by several mechanisms. A few examples of such mechanisms are decreasing synthesis of HMG-CoA which controls cholesterol synthesis (hydroxy-3-methyglutaryl coenzyme A reductase), suprressing synthesis of new LDL receptors in the cells, activating the enzyme acyl-coenzyme A cholesterol acltransferase which helps esterify free cholesterol to cholesterol ester.
If LPLD is present
There will be no lipoprotein lipase present on the endothelial cells. As such, mature chylomicrons and VLDLs containing a high percentage of triglyceride will accumulate in the blood stream, resulting to hypertriglyceridemia. Adipose and muscle tissues will be unable to utilize the triglycerides from the chylomicrons and VLDLs, thus they will have to get their energy from somewhere else (i.e carbohydrate metabolism).
Additionally, VLDL cannot be broken down to form IDL or LDLs, thus cholesterol cannot be utilized by the cells for cell membrane and hormone synthesis, leading to more problems.
Additionally, VLDL cannot be broken down to form IDL or LDLs, thus cholesterol cannot be utilized by the cells for cell membrane and hormone synthesis, leading to more problems.
Majority of those who have LPLD develop symptoms before age 10, experiencing episodes of severe pain and are not able to utilize the calories needed to gain weight and grow as normal.
Common clinical features are abdominal pain caused by frequent recurring pancreatitis, eruptive xanthomas, lipaemia and atherosclerosis.
Psychoneurological manifestations can also be seen often - paresthesias (tingling sensation) of hands, peripheral neuropathies (damage of nerves in periphery system) and reversible form of dementia.
Some of the common symptoms and reasons behind such symptoms are listed in the table below.


Someone with xanthomas on her eyelids, which could potentially impair her eyesight

Left tubes ( In both pictures ) showed blood sample with a clear serum, while the right tubes ( In both pictures ), show blood sample with milky white appearance due to high lipid content, this is also know as lipemia.
LPL activity
A blood test can be taken to measure the acitivity of lipoprotein lipase, where chylomicrons will be emulsified. If lipoprotein lipase activity is low, there will be a lactescent (milky) appearance in the fasting plasma due to the accumulation of triglycerides.

Genetic Testing
A blood or saliva sample is obtained and the LPL gene is checked for any abnormalities. As LPLD is an inherited disease, kin of a person suffering from LPLD has a 25% chance of getting the disease, and 50% chance of being a carrier.

Blood of a person with hyperlipidemia (possibly due to LPLD)
Furthermore, a person can be diagnosed with LPLD if lipoprotein lipase activity is low when the blood plasma and apoprotein C-II are of normal levels.
There are two ways to measure LPL enzyme activity:
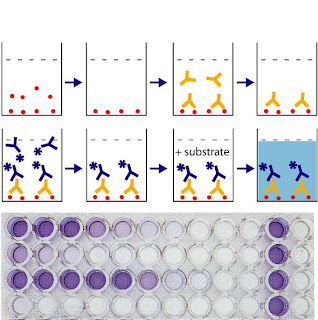
1) Using post-heparin plasma, where 60U/kg body wt. of herparin is administered to release the enzyme into the blood stream. ELISA is used to detect the activity.
2) Using biopsies of apidpose tissue

A picture briefly depicting the steps of ELISA (Enzyme Linked ImmunoSorbent Assay)
Carriers of this mutation generally have a lower lipoprotein lipase activity than an average person, and this can be confirmed through genetic testing.
Genetic Testing
A blood or saliva sample is obtained and the LPL gene is checked for any abnormalities. As LPLD is an inherited disease, kin of a person suffering from LPLD has a 25% chance of getting the disease, and 50% chance of being a carrier.
Present treatment available
Currently, the most effective treatment for symptoms such as pancreatitis is a lipid-lowering therapy by restriction of fat intake (maximum of 20 grams of fat per day). Animal and vegetable fat intake has to be lowered to 15% of calories with the purpose of lowering plasma triglyceride levels to below 1000 or 2000 mg/dl. Food containing medium chain triglycerides, although having a compromised taste, can be taken as they are not transported by chylomicrons.
To ensure that patients have sufficient nutrients, it is recommended for them to take fat-soluble vitamins A, D, E, and K and mineral supplements.
It is advised to avoid alcohol and medication (such as estrogens, diuretics, beta-blockers) as it may increase the concentration of triglyceride in plasma.
Triglyceride levels will automatically rise in pregnant ladies, hence those with LPLD have to pay especially close attention to their blood plasma triglyceride levels and follow a very strict diet to lower the risk of pancreatitis which can be threatening to both themselves and the baby they carry.
Genetic counselling is available for parents whose children are diagnosed with LPLD and counselling can cover
- Information about LPLD
- Evaluation of family medical history to analyse the chance of occurrence of LPLD in other family members
- Benefits of medium-chain triglyceride fats
- Importance of the restriction of fats - both saturated and unsaturated
- How fat-lowering drugs are ineffective
- Avoidance of agents (alcohol and drugs) that cause increase in triglyceride levels
- Strategies for extremely strict diets during pregnancy
Possible treatment
Another treatment, possibly a cure for this hereditary disease, is gene therapy.
Alipogene Tiparvovec (also known as Glybera) is the first gene therapy treatment that has been approved by regulatory authorities in the western world to treat LPLD. The vector contains the human lipoprotein lipase gene variant LPLS447X and has the protein shell that is dervied from adeno-associated virus serotype 1 (AAV1). Glybera is administered through injection into muscle groups, where healthy functional LPL enzymes are made.

This gene therapy has undergone 3 clinical tests in Netherlands and Canada, indicated positive results and has no side effects. The last clinical study conducted in Canada has shown that the effect of breaking down chylomicrons lasted for a significant period of time when a LPLD patient has been treated with one administration of Glybera.
More clinical studies are needed to determine the effectiveness and safety of this treatment.
Additionally, a possible prevention of this disease is germ-line therapy, whereby the human sperms or eggs are genetically altered to prevent the occurrence of the disease. However this has a very small chance of happening due to many ethical issues.
Story of someone who has LPLD

Her name is Jill Prawer and she was diagnosed with LPLD in 1964.
Jill’s parents would always pacify her with sweets and chocolate whenever she cries when she was a baby. But after consuming such treats, she would break out in yellow spots. This was brought to her parent's attention and she was sent to the hospital . At the age of 2, Jill was being diagnosed with Familial LDLP. Since then, her life took a dramatic change where she had to follow a very strict low fat diet plan.Travelling and eating out was a chore to her. In the sixties, there was a lack of nutritional labelling on food, thus it was difficult to make wise consumer choices. As such, she suffered from extreme depression in her late twenties and it took her quite a while to get out of it.
Another treatment, possibly a cure for this hereditary disease, is gene therapy.
Alipogene Tiparvovec (also known as Glybera) is the first gene therapy treatment that has been approved by regulatory authorities in the western world to treat LPLD. The vector contains the human lipoprotein lipase gene variant LPLS447X and has the protein shell that is dervied from adeno-associated virus serotype 1 (AAV1). Glybera is administered through injection into muscle groups, where healthy functional LPL enzymes are made.
How gene therapy works
More clinical studies are needed to determine the effectiveness and safety of this treatment.
Additionally, a possible prevention of this disease is germ-line therapy, whereby the human sperms or eggs are genetically altered to prevent the occurrence of the disease. However this has a very small chance of happening due to many ethical issues.
Story of someone who has LPLD
Her name is Jill Prawer and she was diagnosed with LPLD in 1964.
Jill’s parents would always pacify her with sweets and chocolate whenever she cries when she was a baby. But after consuming such treats, she would break out in yellow spots. This was brought to her parent's attention and she was sent to the hospital . At the age of 2, Jill was being diagnosed with Familial LDLP. Since then, her life took a dramatic change where she had to follow a very strict low fat diet plan.Travelling and eating out was a chore to her. In the sixties, there was a lack of nutritional labelling on food, thus it was difficult to make wise consumer choices. As such, she suffered from extreme depression in her late twenties and it took her quite a while to get out of it.
Jill also had permanent diabetes due to the raised levels of triglyceride in her body after giving birth to three children. Due to the rareness of her condition, Jill felt out of place and lonely.
==========================================
Summary of Research Article
Triglyceride-Induced Diabetes Associated with Familial
Lipoprotein Lipase Deficiency
Introduction
This study is about the correlation between raised
plasma triglycerides (TGs) and nonesterified fatty acids (NEFA) concentrations
and insulin-resistant diabetes, based on a family consisting of 3 daughters.
The father had moderate hypertriglyceridemia, but apart from that, was still healthy.
The mother had a history of recurrent pancreatitis but does not have
hypertriglyceridemia or type 2 diabetes. The two older daughters have
hyperchylomicronemia, who 1-2 years later, developed early onset insulin
resistant diabetes. The youngest sister is unaffected.
Medical therapy (low-calorie low-fat diet, anti-diabetic
medication, human insulin) was done but did not control glycemia or reduce
plasma TGs.
In the end, modified biliopancreatic division (BPD)
was performed, causing lipid mal-absorption.
Techniques Used
Many tests were done before and after the surgery.
Enzymatic colorimetric methods were used to detect
NEFA and TG levels. Polyethylene glycol precipitation kit and cholesterol
oxidase peroxidase antiperoxidase method is used to detect HDL and HDL3
cholesterol. Single radial immunodiffusion technique is used to detect
apolipoprotein C-II. Beckman BUN Analyzer II is used to detect urinary nitrogen
while near-infrared reflectance analysis is used to detect stool fat, starch
and nitrogen. Post-heparin blood with lipolytic activity was also obtained using
intravenous sampling catheter and catalytic LPL assay is done to measure LPL
activity using the amount of [3H]oleic from LPL-catalyzed hydrolysis that
breaks down intralipid emulsion containing the [3H]triolein label.
Molecular analysis using GC-clamped polymerase chain
reaction and denaturing gradient gel electrophoresis (DGGE) is done to detect
any change in nucleotide sequence of the exons in the LPL gene of the family
members.
Results
It is found that the two affected sisters have
inherited their father’s silent mutation on exon 4 and their mother’s silent
mutation on exon 8. The youngest inherited the same mutational pattern on exon
4 and her mother’s nonsense mutation on exon 9.
LPL activity has been found to be lower in the two
girls with hypochylomicronemia. However BPD causes lipid mal-absorption, which
leads to the following:
The fasting levels of TGs, NEFAs, and cholesterol
declined drastically post-surgery. Fasting plasma insulin concentrations were
initially 6-8 times higher than other family members in the sisters but
returned to normal after surgery. Glycemic control was normalized.
Near-infrared reflectance analysis also showed that 83-92% of ingested fat was
recovered in the feces of the sisters. Insulin sensitivity was initially low
but increased drastically after surgery. Weight gain is also observed in both
girls. Diabetes is thus well controlled
without any dietary restrictions.
Using the oral glucose tolerance test, plasma TGs were
closely correlated with both plasma glucose and insulin concentrations,
indicating that insulin-resistant diabetes can be caused by high levels of TGs.
Discussion/
Conclusion
The silent mutations in both parents do not cause
structural alteration of the gene product, though it may be accountable for the
reduced amount of LPL protein which is responsible for breaking down TG in
blood. However the termination mutation in exon 9 causes an increase in LPL
activity and HDL cholesterol while lowering TG concentration in the
bloodstream. Rare factors that are difficult to detect such as intron mutations
may be the reason why it does not occur in the two children and mother.
It has been observed that reduced LPL activity has
caused higher levels of plasma TGs and high levels of NEFAs may have caused
insulin resistance. Both sisters are cured of diabetes when their TG and NEFA
levels decreased and their glucose uptake increased to that of their father’s.
==========================================
References
Bruno M. J. (2010). Gene Therapy Coming of Age - Prevention of Acute Pancreatitis In Lipoprotein Lipase Deficiency Throgh Alipogene Tiparvovec. Available at: <http://www.uniqure.com/uploads/Publications/GL0011_clinical_2010.pdf> Last accessed 17 July 2013.David C. D. (22 March 2013). Familial lipoprotein lipase deficiency.Available: http://www.nlm.nih.gov/medlineplus/ency/article/000408.htm. Last accessed 17 July 2013.
Genetics Home Reference. (January 2008). Familial lipoprotein lipase deficiency. Available at: <http://ghr.nlm.nih.gov/condition/familial-lipoprotein-lipase-deficiency> Last accessed 17 July 2013.
Genetics Home Reference. (January 2008). LPL. Available at: <http://ghr.nlm.nih.gov/gene/LPL> Last accessed 17 July 2013.
John D B. (15 December , 2011). Familial Lipoprotein Lipase Deficiency.Available: http://www.ncbi.nlm.nih.gov/books/NBK1308/. Last accessed 17 July 2013.
Kenya Forensics Online Resource (15 May 2012) Image of ELISA. Available at: <http://kenyaforensics.blogspot.sg/2012/05/application-of-elisa-in-forensic.html> Last accessed 17 July 2013.
Kori J. K, RN, MSN, Greg B. (N.D). Understanding the Essentials of Blood Lipid Metabolism. Available: <http://www.medscape.com/viewarticle/451762_5>. Last accessed 23 July 2013.
Meghan E. C. (N.D). Genetic Counseling. Available: http://web.ornl.gov/sci/techresources/Human_Genome/medicine/genecounseling.shtml. Last accessed 17 July 2013.
Mingrone G., Henriksen F. L., Greco A. V., Krogh L. N., Capristo E., Gastaldelli A., Castagneto M., Ferrannini E., Gasbarrini G., Beck-Nielsen H. (1999) Triglyceride-Induced Diabetes Associated with Familial Lipoprotein Lipase Deficiency. Diabetes 48:1258–1263. Available at: <http://diabetes.diabetesjournals.org/content/48/6/1258.long> Last accessed 22 July 2013.
National Human Genome Research Institute Organization. (30 April 2013). Frequently Asked Questions About Genetic Counseling. Available: http://www.genome.gov/19016905. Last accessed 17 July 2013.
NORD. (25 April 2008). National Organization for Rare Disorders, Inc..Available: http://icmmt.alere.com/kbase/nord/nord474.htm. Last accessed 17 July 2013.
Palmer M., University of Waterloo (n.d.) Formation of chylomicrons. Available at: <http://watcut.uwaterloo.ca/webnotes/Metabolism/page-10.1.2.html> Last accessed 17 July 2013
Simon P., Ulrich F. W., Zimmer K., Koch H. G., Lerch M. M.(2001). Acute and Chronic Pnacreatitis in paitents with inborn errors of metabolism. Available at: <http://www.medizin.uni-greifswald.de/gastro/Veroeffentlichungen/29.pdf> Last accessed 17 July 2013.
The AOCS Lipid Library (n.d.) Cholesterol and Cholesterol Esters. Available at: <http://homepage.smc.edu/wissmann_paul/anatomy2textbook/1cholesterol.html> Last accessed 22 July 2013.
uniQure. (2012). Alipogene Tiparvovec (Glybera ®)- a Gene Therapy for LPLD. Available: http://www.uniqure.com/uploads/Glybera%202pp%20Factsheet_lr.pdf. Last accessed 17 July 2013.
Walid Aziz Basharyar (9 May 2008) Lipoprotein Physiology: Overview (1/4). Available at: <http://www.youtube.com/watch?v=4D6CqHpvVz8> Last accessed 22 July 2013
Walid Aziz Basharyar (9 May 2008) Lipoprotein Physiology: Chylomicron (2/4). Available at: <http://www.youtube.com/watch?v=xAqL9fLwnDs&hl=en-GB&gl=SG> Last accessed 22 July 2013
Zhang X., Rong Qi, X. Xian, Fei Yang, Blackstein M., Deng X., Fan J., Ross C., Karasinska J.,Hayden M.R., Liu G. (2008) Spontaneous Atherosclerosis in Aged Lipoprotein Lipase–Deficient Mice With Severe Hypertriglyceridemia on a Normal Chow Diet. Circulation Research. 102: 250-256. Available at: <http://circres.ahajournals.org/content/102/2/250.full> Last accessed 17 July 2013.